While this conclusion was something more or less reachable with previous data (see
HERE for example), a new study adds some fine detail for us to reconstruct the paleohistory of this major Eurasian lineage.
Peter A. Underhill et al., The phylogenetic and geographic structure of Y-chromosome haplogroup R1a. EJHG 2014. Pay per view → LINK [doi:10.1038/ejhg.2014.50]
Abstract
R1a-M420 is one of the most widely spread Y-chromosome haplogroups; however, its substructure within Europe and Asia has remained poorly characterized. Using a panel of 16 244 male subjects from 126 populations sampled across Eurasia, we identified 2923 R1a-M420 Y-chromosomes and analyzed them to a highly granular phylogeographic resolution. Whole Y-chromosome sequence analysis of eight R1a and five R1b individuals suggests a divergence time of ~25 000 (95% CI: 21 300–29 000) years ago and a coalescence time within R1a-M417 of ~5800 (95% CI: 4800–6800) years. The spatial frequency distributions of R1a sub-haplogroups conclusively indicate two major groups, one found primarily in Europe and the other confined to Central and South Asia. Beyond the major European versus Asian dichotomy, we describe several younger sub-haplogroups. Based on spatial distributions and diversity patterns within the R1a-M420 clade, particularly rare basal branches detected primarily within Iran and eastern Turkey, we conclude that the initial episodes of haplogroup R1a diversification likely occurred in the vicinity of present-day Iran.
This case, as well as many others, including that of its close relatives R1b and Q, illustrate why frequency is not the same as origin, which can only be inferred (if at all) by studying the hierarchical diversity of the lineage. These three lineages for example, must have spread from West Asia but they are relatively less important in numbers in that region today, overshadowed by other lineages, notably J. Instead their derived branches had major impacts in other regions (Europe, South and Central Asia, Siberia and America).
Frequencies of the main lineages
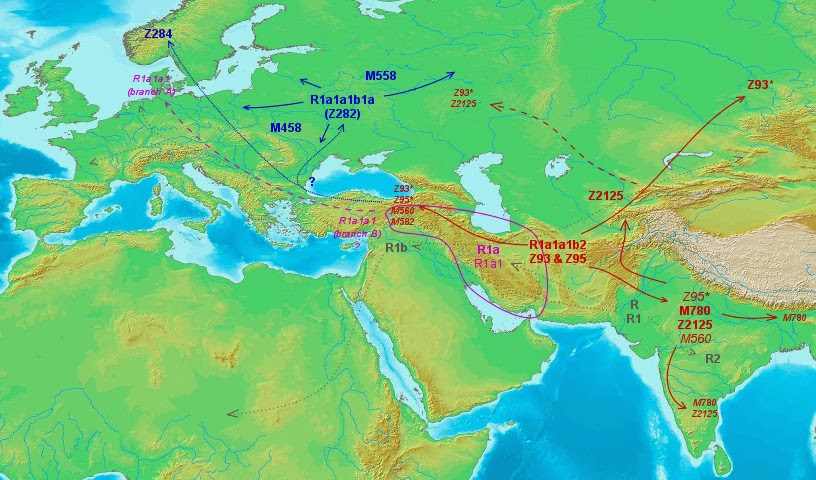
There are two main sub-lineages of R1a, which according to the current ISOGG tree version (maybe to be refitted after this study?) are known as R1a1a1b2 (Z93) and R1a1a1b1a (Z282). The first one is essentially Asian (with greatest frequencies in South and Central Asia, where it includes >98% of all R1a individuals) wile the latter is almost exclusively European (notably Eastern European but with a distinct branch in Scandinavia, encompassing together >96% of R1a individuals in Europe).
These maps give us a quite decent glimpse of the main scatter patterns of R1a but alone they can't inform us of its origins. For that we have to look at the detailed tree and the relationship of its samples with geography.
Origins and distribution of R1a
As mentioned above, the authors conclude that R1a and R1a1 must come from Iran, where the greatest basal diversity is:
To infer the geographic origin of hg R1a-M420, we identified populations harboring at least one of the two most basal haplogroups and possessing high haplogroup diversity. Among the 120 populations with sample sizes of at least 50 individuals and with at least 10% occurrence of R1a, just 6 met these criteria, and 5 of these 6 populations reside in modern-day Iran. Haplogroup diversities among the six populations ranged from 0.78 to 0.86 (Supplementary Table 4). Of the 24 R1a-M420*(xSRY10831.2) chromosomes in our data set, 18 were sampled in Iran and 3 were from eastern Turkey. Similarly, five of the six observed R1a1-SRY10831.2*(xM417/Page7) chromosomes were also from Iran, with the sixth occurring in a Kabardin individual from the Caucasus. Owing to the prevalence of basal lineages and the high levels of haplogroup diversities in the region, we find a compelling case for the Middle East, possibly near present-day Iran, as the geographic origin of hg R1a.
Between these top tier nodes (R1a and R1a1) and the two most common sublineages described above, this study only found one paragroup represented: R1a1a1* (M417). This should be an important step in the analysis but the researchers prefer to remain silent on it. Why? I guess that the reason is that it is complicated to analyze and reach to sound conclusions.
I spent some time today looking at the haplotypes of this paragroup mentioned in the study and I could not reach a conclusion either: the majority of the sequences are from Europe and all them (excepting a highly derived Norwegian line and including a low derived Iranian one) seem to derive from a North German haplotype. I call this group "branch A".
However there is at least one West Asian sequence (from Turkey) which seems independent ("branch B"), while an Indian and the already mentioned Norwegian sequence could derive from either one. So my impression is that there is an specifically North European "branch A" but also some other stuff with West Asian centrality ("branch B") within this key paragroup.
Guess that I could say a lot more about not being able to say much more on this key intermediate step but, synthetically there are two options among which I can't decide:
- Branch A went back to West Asia from where it spread again to Eastern Europe and Central South Asia.
- Branch B is actually at the origin of the two derived and highly spread subhaplogroups.
Whatever the case I understand that there are good reasons to think that these spread first from West Asia, at the very least Z93 and very likely also Z282.
R1a1a1b2 (Z93)
There is nothing European in this lineage: only some lesser terminal branches at the Southern Urals, roughly where the Kurgan phenomenon began some 6000 years ago.
This detail is indeed remarkable because, if, as often argued, R1a or some of its subclades spread from there, we should expect at least some basal diversity being retained. Instead all we see are some highly derived branches. So the main conclusion must be that the expansion of R1a does not seem related to the Kurgan phenomenon, except maybe in some secondary instances.
As mentioned before, this lineage is Central and South Asian and comprises the vast majority of R1a in those two regions.
In essence we can say that:
- Z93* has three apparent distinct branches stemming from West Asia (incl. Caucasus) and another one from South Asia/Altai (1).
- Z95* has two apparent distinct branches:
- A small one with presence in West Asia and Southern Europe
- Another one (pre-M780?) stemming from South or West Asia
- M780 has clear origins in South Asia (incl. most Roma lineages)
- Z2125 also appears to originate in South Asia, even if it has a greater spread outside it, notably to Central Asia
- M580 and M582 appear related and surely originated in West Asia
Weighting them:
- Z95:
- West Asia: 2
- South Asia: 2
- West/South Asia: 1
Therefore the origin of Z95 should be though as West-South Asian but undecided between either region. Say Afghanistan for example.
- Z93:
- West Asia: 3
- West/South Asia: 1 (Z95)
- South Asia/Altai: 1
In this case I would say that West Asia is almost certainly the origin, although tending to Central/South Asia. For example: Iran again.
So, regardless of whether the previous stage (M417) represents a stay in West Asia or a back-migration from Europe into West Asia, West Asia is clearly at the origin of Z93. It does not represent any Kurgan migration but an Asian phenomenon with origins towards the West (around Iran).
R1a1a1b1a (Z282)
On first sight this European sublineage seemed quite simpler: it is obvious that the bulk of it spread from Eastern Europe. However, when we look at
the haplotype network, we cannot confirm this pattern for the Norwegian or Scandinavian haplogroup Z284, which is only linked to the rest via some South European and West Asian samples.
So my conclusion must be that Z282 experienced a main expansion from Eastern Europe but only into Eastern and Central Europe and that the Scandinavian variant almost certainly represents another flow within this haplogroup, with the knot being in West Asia.
Anyhow the main East and Central European expansion seems true. For some reason it is not centered in any obvious prehistorical locality, as could be the Volga or maybe Ukraine, but instead its center is further North around Smolensk.
Overall reconstruction of the spread of R1a
With all the previous analysis I made this map, which also shows in discrete gray color the general pattern of expansion of haplogroup R:
We have an expansion of R into South Asia and Western Eurasia (incl. Central Asia) and even into parts of Africa (R1b-V88) from apparent South Asian (R, R1 and R2) and West Asian (R1a, R1b) origins. Related lineages Q and P* could also be integrated into this pattern of expansion but I did not want to overload the map with too many details.
There is some uncertainty regarding the North European branches of R1a but otherwise the pattern seems quite clear.
On these North European branches, I must say that they remind me of other odd lineages with similar geography: R1b-U106, I1-M253 and I2a2-M223. With the likely exception of R1b-U106 neither appears to have experienced any significant re-expansion since their arrival to that corner of the World, however they do seem to survive pretty well in it.
Time frame?
Finally we seem to be entering the age of full Y chromosome sequencing and a more serious molecular clock based on it. As I have explained on other occasions (
for example), the human Y chromosome is large enough to experience mutations almost every single generation, what should provide a decent molecular clock, unlike the very rough approximations used in the past.
However the issue of correct calibration remains open. As you surely know the academy is slow to incorporate the most recent evidence, especially from fields distinct to their specialty. Hence I do not expect them to calibrate based on the obvious fact that age(CF) or at least age(F)=100,000 years. They are probably still stuck in old concepts of a "recent" out-of-Africa migration c. 60 or at most 80 Ka ago, as well as the usual Pan-Homo spilt under-estimates
I must reckon in any case that I had not enough time to study this matter in depth yet, so the previous observation is rather my idea of what to expect.
In any case in this study the authors resorted to full Y chromosome to calculate their age estimates and I applaud them for doing so. As apparent in fig. 5, all R1 derived sequences have approximately the same number of accumulated SNPs, what in principle allows for a perfected molecular clock, assuming it is well calibrated.
Their estimate is as follows:
A consensus has not yet been reached on the rate at which Y-chromosome SNPs accumulate within this 9.99Mb sequence. Recent estimates include one SNP per: ~100 years,⁵⁸ 122 years,⁴ 151 years⁵ (deep sequencing reanalysis rate), and 162 years.⁵⁹ Using a rate of one SNP per 122 years, and based on an average branch length of 206 SNPs from the common ancestor of the 13 sequences, we estimate the bifurcation of R1 into R1a and R1b to have occurred ~25,100 ago (95% CI: 21,300–29,000). Using the 8 R1a lineages, with an average length of 48 SNPs accumulated since the common ancestor, we estimate the splintering of R1a-M417 to have occurred rather recently, B5800 years ago (95% CI: 4800–6800). The slowest mutation rate estimate would inflate these time estimates by one third, and the fastest would deflate them by 17%.
The references correspond to (4) Poznick 2013, (5) Francalacci 2013, (58) Xue 2009 and (59) Méndez 2013. This last is the Anzick study, of which at the very least we can say that they had a real calibration point in the ancient Amerindian DNA. It is also the one which provides the longest mutation rate.
Considering that Xue 2009 is "old" (for this avant-guard aspect of this pretty young science), I find their choice of the Poznick rate quite a bit conservative. The Francalacci rate is the intermediate one of the three "recent" papers referenced and it is also quite close to the calibrated Méndez rate.
Personally I would choose the later without a second thought. As long as CF ends up being younger than 100 Ka, it is positively too conservative anyhow.
Using the Méndez (Anzick-calibrated) rate of 162 years per SNP, I get the following corrected estimates:
- R1a/R1b split (R1 node): 33,000 years ago (CI: 26.0-42.5 Ka)
- R1a-M417 node: 7,700 years ago (CI: 6.4-9.0 Ka)
These seem fair enough to me, judging on the fact that the core R1a expansion seems to originate in West Asia (at the very least for the South/Central Asian branch), what fits much better with a Neolithic frame than with the Kurgan one.
In my understanding the R1 node is actually c. 48 Ka old (R1b: c. 34 Ka.), what, apportioning, yields a date of c. 11.2 Ka for the R1a-M-417 node.
Update (Mar 31):best possible molecular clock estimates for R1:
Follows fig. 5 of Underhill et al. 2014, annotated by me in red and purple colors:
If I'm correct, then the expansion of
R1b in Europe still corresponds in rough terms to the Magdalenian period or, more generally, the late Upper Paleolithic. This does not mean that it remained that way forever (it may well have been reshuffled later on: in the Epipaleolithic, Neolithic and Chalcolithic) but it seems to be the time-frame of its main expansion when the main lineages got established, whatever happened to them later on.
I know well that so far ancient DNA for this lineage remains to be found and that the dominant haplogroup among known Epipaleolithic hunter-gatherers was (for all we know) I2a. However this is what the refined full Y chromosome sequence molecular clock, properly calibrated according to the archaeological evidence for the settling of Asia by H. sapiens, has to say. If you wish to dismiss this and use another estimate instead, that's always up to you. I just hope that you know what you're doing.
Anyhow, if I am correct, then the expansion of R1a is neither Chalcolithic nor Neolithic but clearly Epipaleolithic. Does it make any sense? I can't say for sure because this period is not so well understood. Whatever the case, is it possible to integrate the key pre-Neolithic
Zarzian culture of the Zagros (
map) in this scheme of things? What about all the other question marks that fill the gaps of our mediocre knowledge of the Mesolithic of West Asia? Or is it the
Balcanic Epigravettian to be blamed instead? Or both?
I really can't say with any certainty at this stage. But I am intrigued indeed.
Update (Mar 31): frequency pie charts of Underhill's data available at Kurdish DNA.
Update (Aug 2015): I must update the frequencies of the various upstream paragroups, in agreement with table S4, because I may have missed some details initially. However the overall tendency is the same.
- R1a* (M420): Italy (1), Turkey East (1), Turkey Cappadocia
(2), UAE (1), Oman (1), Iran (set 2) (2), Iran NE (1), Iran South (5),
Iran North (5), Azeris-Iran (5).
- R1a1* (SRY10831.2): Iran (set 2) (1), Iran NE (1), Iran South (2), Iran North (1), Kabardin (1). In addition it has more recently been found in two Epipaleolithic Eastern Europeans (EHG), from Karelia (Haak 2015) and Smolenskaya Oblast (Chekunova 2014).
- Ra1a1a1* (M417): Ireland (1), Netherlands (3), Norway (1), South Sweden (1), Germany (1),
Estonia (1), Hungary (1), Turkey East (Kurds) (1), Iran (set 3) (1),
India South (1).



















{kind=link}