Showing posts with label Genetics. Show all posts
Showing posts with label Genetics. Show all posts
April 28, 2018
Video: Do genes make you fat?
I don't usually deal with the medical aspects of genetics but this conference by Giles Yeo is so enticing and clarifying that I believe it deserves an entry here:
March 31, 2018
A new genetics blog in Spanish

Wilhelm H > DNA and Genealogy, by some guy called Wilhelm Halys, whom I know from Facebook.
His very first post on endogamy using RootsFinder seems very interesting, even if I have to admit I don't understand it well because I'm unfamiliar with this program. But that's good because it indicates novelty, fresheness and stuff to learn.
Hope you also find interesting, assuming you can read Spanish or use a translator.
November 2, 2015
Selection against Neanderthal introgression?
Quickies
A couple of papers have been pre-published these days discussing the apparent selection against most (but not all) of the Neanderthal inheritance among modern ex-Africa humans.
Ivan Juric, Simon Aeschbacher &
Graham Coop, The Strength of Selection Against Neanderthal
Introgression. BioRxiv 2015 (pre-pub). Freely accessible → LINK [doi:
http://dx.doi.org/10.1101/030148]
AbstractHybridization between humans and Neanderthals has resulted in a low level of Neanderthal ancestry scattered across the genomes of many modern-day humans. After hybridization, on average, selection appears to have removed Neanderthal alleles from the human population. Quantifying the strength and causes of this selection against Neanderthal ancestry is key to understanding our relationship to Neanderthals and, more broadly, how populations remain distinct after secondary contact. Here, we develop a novel method for estimating the genome-wide average strength of selection and the density of selected sites using estimates of Neanderthal allele frequency along the genomes of modern-day humans. We confirm that East Asians had somewhat higher initial levels of Neanderthal ancestry than Europeans even after accounting for selection. We find that there are systematically lower levels of initial introgression on the X chromosome, a finding consistent with a strong sex bias in the initial matings between the populations. We find that the bulk of purifying selection against Neanderthal ancestry is best understood as acting on many weakly deleterious alleles. We propose that the majority of these alleles were effectively neutral-and segregating at high frequency-in Neanderthals, but became selected against after entering human populations of much larger effective size. While individually of small effect, these alleles potentially imposed a heavy genetic load on the early-generation human-Neanderthal hybrids. This work suggests that differences in effective population size may play a far more important role in shaping levels of introgression than previously thought.
Kelley Harris & Rasmus Nielsen, The Genetic Cost of Neanderthal Introgression. BioRxiv 2015 (pre-pub). Freely accessible → LINK [doi: http://dx.doi.org/10.1101/030387]
AbstractApproximately 2-4% of the human genome is in non-Africans comprised of DNA intro- gressed from Neanderthals. Recent studies have shown that there is a paucity of introgressed DNA around functional regions, presumably caused by selection after introgression. This observation has been suggested to be a possible consequence of the accumulation of a large amount of Dobzhansky-Muller incompatibilities, i.e. epistatic effects between human and Neanderthal specific mutations, since the divergence of humans and Neanderthals approx. 400-600 kya. However, using previously published estimates of inbreeding in Neanderthals, and of the distribution of fitness effects from human protein coding genes, we show that the average Neanderthal would have had at least 40% lower fitness than the average human due to higher levels of inbreeding and an increased mutational load, regardless of the dominance coefficients of new mutations. Using simulations, we show that under the assumption of additive dominance effects, early Neanderthal/human hybrids would have experienced strong negative selection, though not so strong that it would prevent Neanderthal DNA from entering the human population. In fact, the increased mutational load in Neanderthals predicts the observed reduction in Neanderthal introgressed segments around protein coding genes, without any need to invoke epistasis. The simulations also predict that there is a residual Neanderthal derived mutational load in non-African humans, leading to an average fitness reduction of at least 0.5%. Although there has been much previous debate about the effects of the out-of-Africa bottleneck on mutational loads in non-Africans, the significant deleterious effects of Neanderthal introgression have hitherto been left out of this discussion, but might be just as important for understanding fitness differences among human populations. We also show that if deleterious mutations are recessive, the Neanderthal admixture fraction would gradually increase over time due to selection for Neanderthal haplotypes that mask human deleterious mutations in the heterozygous state. This effect of dominance heterosis might partially explain why adaptive introgression appears to be widespread in nature.
September 5, 2015
Selection of dominant alleles vs recessive ones in population bottlenecks
Quantity over quality series
A technical study that however gives an interesting glimpse on the complex but generally reductive genetic effect of bottlenecks such as the Out-of-Africa migration of humankind is out in PLoS Genetics.
Daniel J. Ballick et al., Dominance of Deleterious Alleles Controls the Response to a Population Bottleneck. PLoS Genetics 2015. Open access → LINK [doi:10.1371/journal.pgen.1005436]
Abstract
Population bottlenecks followed by re-expansions have been common throughout history of many populations. The response of alleles under selection to such demographic perturbations has been a subject of great interest in population genetics. On the basis of theoretical analysis and computer simulations, we suggest that this response qualitatively depends on dominance. The number of dominant or additive deleterious alleles per haploid genome is expected to be slightly increased following the bottleneck and re-expansion. In contrast, the number of completely or partially recessive alleles should be sharply reduced. Changes of population size expose differences between recessive and additive selection, potentially providing insight into the prevalence of dominance in natural populations. Specifically, we use a simple statistic,, where xi represents the derived allele frequency, to compare the number of mutations in different populations, and detail its functional dependence on the strength of selection and the intensity of the population bottleneck. We also provide empirical evidence showing that gene sets associated with autosomal recessive disease in humans may have a BR indicative of recessive selection. Together, these theoretical predictions and empirical observations show that complex demographic history may facilitate rather than impede inference of parameters of natural selection.
Dominance has played a central role in classical genetics since its inception. However, the effect of dominance introduces substantial technical complications into theoretical models describing dynamics of alleles in populations. As a result, dominance is often ignored in population genetic models. Statistical tests for selection built on these models do not discriminate between recessive and additive alleles. We show that historical changes in population size can provide a way to differentiate between recessive and additive selection. Our analysis compares two sub-populations with different demographic histories. History of our own species provides plenty of examples of sub-populations that went through population bottlenecks followed by re-expansions. We show that demographic differences, which generally complicate the analysis, can instead aid in the inference of features of natural selection.
Fig 3. The BR statistic at the time of observation.
ABOVE: At the time of observation tobs, the value of BR(tobs) is plotted as a function of the average strength of selection s and dominance coefficient h. Dominance coefficients appear as solid lines with fully recessive selection (h = 0) at the top and purely additive selection (
 ) at the bottom. For strong selection BR → 1 due to the rapid transient response. For weak selection BR → 1 due to the nearly neutral insensitivity to the bottleneck. For some intermediate dominance coefficient hc, a critical value occurs (hc ~ 0.25 in the example shown, but explored more generally in S1 Text) where additive and recessive effects cancel, yielding BR(hc) ~ 1. A low intensity bottleneck (IB = 0.05) is shown, with parameters 2N0 = 20000, 2NB = 2000, TB = 100, and tobs = 1000. BELOW: The same range of parameters is plotted for a realistic demographic model of the Out of Africa event comparing Africans and Europeans [48], where BR = 〈x〉African/〈x〉European. The European bottleneck has estimated intensity IB ~ 𝒪(0.5), an order of magnitude stronger than the simple bottleneck above, allowing for potentially observable deviations from BR ~ 1 if a large fraction of analyzed variants act recessively with h < hc ~ 0.25. ) at the bottom. For strong selection BR → 1 due to the rapid transient response. For weak selection BR → 1 due to the nearly neutral insensitivity to the bottleneck. For some intermediate dominance coefficient hc, a critical value occurs (hc ~ 0.25 in the example shown, but explored more generally in S1 Text) where additive and recessive effects cancel, yielding BR(hc) ~ 1. A low intensity bottleneck (IB = 0.05) is shown, with parameters 2N0 = 20000, 2NB = 2000, TB = 100, and tobs = 1000. BELOW: The same range of parameters is plotted for a realistic demographic model of the Out of Africa event comparing Africans and Europeans [48], where BR = 〈x〉African/〈x〉European. The European bottleneck has estimated intensity IB ~ 𝒪(0.5), an order of magnitude stronger than the simple bottleneck above, allowing for potentially observable deviations from BR ~ 1 if a large fraction of analyzed variants act recessively with h < hc ~ 0.25. |
I emphasize from the erudite legend:
The European bottleneck has estimated intensity IB ~ 𝒪(0.5), an order of magnitude stronger than the simple bottleneck above.
Although Europeans are used for reference this bottleneck and the corresponding accumulation of deleterious alleles is the same for all non-Africans.
June 29, 2014
Pan-Homo split: 11-17 million years ago
Chimpanzee mutation rate is largely determined by fathers' age and, overall, implies a Pan-Homo divergence rate of ~13 million years (95% CI: 11-17 Ma), about double than usually assumed by conservative scholastic inertia.
Oliver Venn et al., Strong male bias drives germline mutation in chimpanzees. Science 2014. Pay per view → LINK [doi:10.1126/science.344.6189.1272]
 |
| cc Matthew Hoelscher |
The focus of this study are the important differences between patrilineal and matrilineal mutation rate depending on the father's age among chimpanzees, notably more biased than among humans. However the resulting estimate for Pan-Homo divergence is not less important because it radically challenges the usual assumptions of 5-7 Ma, repeated once and again in molecular clock estimates, which are based on studies that are already quite obsolete.
In the studied captive population of Western chimpanzees 30 out of 35 mutations happened in the paternal lineage, and these increase with the father's age. No effect could be attributed to maternal age or familiar peculiarities.
Interestingly most of these patrilineal mutations happen near the telomeres, an effect not seen in female line mutations.
Owing to this gender bias, the mutation rate of the X chromosome among chimpanzees is 74% that of autosomal DNA (in humans: 85%).
The gender bias in mutation rate and its differential with humans is attributed to differences in mating systems among great apes, with chimpanzees having the greatest competition among males, what is reflected in testicle size. They predict that gorillas (who experience less competition between males) will show less patrilineal mutation rate bias than humans and chimpanzees.
This is probably the more synthetic paragraph from the study:
Under a model in which the mutation rate increases linearly with parental age, the rate of neutral substitution is the ratio of the average number of mutations inherited per generation to the average parental age. We predict the neutral substitution rate to be ~0.46 × 10−9 per base pair (bp) per year in chimpanzees, compared to estimates in humans of ~0.51 × 10−9 bp−1 year−1 (9). These results are consistent with near-identical levels of lineage-specific sequence divergence (12) but surprising given the differences in paternal age effect. In the intersection of the autosomal genome accessible in this study and regions where human and chimpanzee genomes can be aligned with high confidence, the rate is slightly lower (0.45 × 10−9 bp−1 year−1) and the level of divergence is 1.2% (13), implying an average time to the most common ancestor of 13 million years, assuming uniformity of the mutation rate over this time (95% ETPI 11 to 17 million years; table S11).
13 million years of the hominid line
This is not at all the first study to highlight the extreme dubiousness of the usual scholastic assumptions regarding the Pan-Homo divergence, which taint so many genetic studies, turning their chronological estimates totally worthless.
In 2010, Wilkinson et al. estimated a Pan-Homo divergence rate of 8-10 Ma. In 2012 Langergraber et al. recalibrated previous studies getting a Pan-Homo divergence bracket of 6.78-13.45 Ma (fig.2), while the divergence from Gorilla would be significantly older: 8.31-20.0.
 |
| Fig. 1 from Langergraber 2012. Legend: Diagram illustrating the branching pattern and timing of the splits between humans, chimpanzees, bonobos, western gorillas, and eastern gorillas. The paler shading indicates the range of split times inferred in this study. Cartoon skulls indicate approximate age of the indicated fossil remains, but do not imply that these fossils were necessarily on those ancestral lineages or that entire crania actually exist for these forms. |
A key fossil affecting this controversy is Sahelanthropus tchadiensis (Toumaï), which has been recently confirmed to be in the human line on several hardly questionable traits and is dated to c. 7 Ma.
A related debate is whether primates in general are much older than usually claimed and lived already in the Jurassic, something suggested by the already mentioned Wilkinson study and also by Heads 2010. Here a major issue is that mainline conservative estimates would have the ancestors of New World monkeys swimming (island hoping) to South America, something that those monkeys (and most other primates) simply will not do. The radiation of primates to South America and possibly also Madagascar is much better explained if these animals could just tree-hop, rather than island-hop to their destinations. However this would demand a radical revision of the usual age estimate for vertebrate radiation, what so far lacks fossil support (but lack of evidence is not evidence of lack, you know: fossil ages can only be taken as terminus ante quem dates and not absolute direct references).
But this is a side question, what really matters to us is that our ancestors split from the chimpanzee line c. 13 Ma (according to this study) and not after 8 Ma in any case (weighting all the evidence). This not just renders most "molecular clock" estimates useless and effectively false (wrong, erroneous, inadequate, misleading, junk, pseudoscientific...) but also help us to rethink our ancestral history in the African savannas since long before we became humans (Homo sp.)
Looking for some ecological context clues, I found this 1996 study by Jean Maley, which shows that Africa was largely humid in the early Miocene (smectite: evidence of water) but that it became increasingly arid towards the middle Miocene (kaolinite: evidence of sand). Up to this key ecological change of the Middle Miocene, the rainforest extended all the way to Egypt and East Africa. This kind of ecology allows for the common ancestor of African great apes to have arrived and first diverged in a jungle-dominated ecology and, later, for the speciation event leading to humans (bipedalism) to have happened as this once widespread jungle became scarcer, yielding to deserts and savanna.
 |
| Sahelanthropus (from fossilized.org) |
It just makes all sense that the evolution of bipedalism was coincident with the vanishing of that originally widespread jungle environment whose dating is of approx. 13 Ma ago. However it must be said that the consolidation of the Sahara only happened much later, c. 7 Ma ago, already approaching the Pliocene.
Regardless of the exact split-time, a big question I have on hominid evolution is how on Earth did our small-brained and small-toothed precursors like Toumaï survive in the open savannas and grasslands without fire nor weapons. Even if they resorted to trees (isolated or in patches) for refuge, there were already felines of the saber-toothed family roaming in Africa and these big cats were no doubt be able to climb on trees and in some cases they have been shown to predate on australopithecines. How could our precursors in the hominin line be able to face this menace without the advantage of speed (as ruminants have) or good defenses? Were their strong forelimbs, together with team action enough to confront the threat of predators? Did they use primitive weapons such as branches and stone throwing?
June 21, 2014
Claim of 13 Ma Pan-Homo split
[Update (Jun 29): new entry on this issue available].
[Update: the origin of this news is Venn 2014 but I could not find the mention of the 13 Ma split initially, as it was not something they underlined at all. I will write something as soon as possible. Thanks to all the people who helped my confused mind].
Live Science reports this week that the divergence of the human and chimpanzee lines may be as old as 13 million years. This is the oldest range of what Langergraber 2012 suggested (8-13 Ma in Fig.1, although in text they wrote "6.8-11.6 Ma") and older than the Wilkinson 2010 estimates (8-10 Ma), and would totally break all the usual "molecular clocks" so extremely abused in human genetics because it is double of the usual scholastic mindless parroting (5-7 Ma, which are necessarily too recent because they do not allow for Sahelanthropus' evolution and not even for bonobo evolution under the protection of the mighty Congo river).
[Update: the origin of this news is Venn 2014 but I could not find the mention of the 13 Ma split initially, as it was not something they underlined at all. I will write something as soon as possible. Thanks to all the people who helped my confused mind].
Live Science reports this week that the divergence of the human and chimpanzee lines may be as old as 13 million years. This is the oldest range of what Langergraber 2012 suggested (8-13 Ma in Fig.1, although in text they wrote "6.8-11.6 Ma") and older than the Wilkinson 2010 estimates (8-10 Ma), and would totally break all the usual "molecular clocks" so extremely abused in human genetics because it is double of the usual scholastic mindless parroting (5-7 Ma, which are necessarily too recent because they do not allow for Sahelanthropus' evolution and not even for bonobo evolution under the protection of the mighty Congo river).
Sadly the article includes no reference to the source, not even the name of the scientists involved, and I could not find it any reference online. For a moment I thought it could be another new study on gender bias in chimpanzee mutation rate (Venn et al. 2014 (ppv)) but after getting a copy it does not seem to have any direct relation.
So I would appreciate if someone can give me a lead on where this claim may come from.
May 24, 2014

May 9, 2013
Echoes from the past (May-9-2013)
I am getting updated with a rather long backlog, so I will speed things up placing here in nearly telegraphic style the informative snippets that require less work. This does not mean that they are less interesting, not at all, just that I have to adapt to that elusive quality of time...
Middle Paleolithic
Toba supervolcano only had short-term climate effect → BBC.
Research on Lake Malawi's sediments shows that the climate-change effect of the catastrophic eruption was limited. Droughts previously believed to be from that period have been revised to be from at least 10,000 years before, corresponding to the end of the Abbassia Pluvial rather than to Toba super-eruption.
Upper Paleolithic
Altai rock art and early astronomy from 16,000 BP → Siberian Times, Daily Mail.
Sunduki (Khakassia), here there are what are surely the oldest rock art of Northern Asia, representing people hunting or interacting among them, which are from just centuries ago, however other petroglyphs are apparently much older like this horse:

Prof. Vitaly Larichev (Institute of Archeology and Ethnography, Russian Academy of Sciences) has detected a whole astronomical structure implemented in the landscape.
He claims to have found 'numerous ancient solar and lunar observatories around Sunduki'.
'This square pattern of stones on the ground shows you the place', he told visiting author Kira Van Deusen. 'I knew there would be an orientation point, but we had to search through the grass for a long time to find it.
'Now look up to the top of that ridge. You see a place where there is a crack between the rocks? If you were here on the summer solstice, you would see the sun rise right there. Or you would if you were here 2,000 years so. Now the timing is slightly differen'.
High on one cliff wall is a rock engraving showing dragon heads in one direction, and snake heads in the other.
'If the sun were shining, we could tell the time,' he said. 'In the morning the shadow moves along the snake's body from his head to his tail, and in the afternoon it comes from the other direction along the dragon.
'From the same observation point you can determine true north and south by sighting along the mountains'.
Neolithic
Vietnam: early cemetery dug in Thahn Hoa → Australian National University.
Some 140 human remains of all ages have been unearthed at the site of Con Co Ngua, estimated to be 6-4000 years old. Cemeteries of this size and age were previously unknown in the region. The site has also revealed a dearth of artifacts.
The people were buried in fetal position with meat cuts of buffalo or deer.
Chalcolithic
India: 4000 y.o. stone tools unearthed in Bhopal (Madhya Pradesh, Narmada river) → India Today.
Details:
- Some of them are decorated with aquatic animals.
- 150x200 m. mound in Birjakhedi
- Terracotta game pieces
- Pottery (incl. jars, pots, dishes)
- Stone and ivory beads
 Bell Beaker rich lady's burial unearthed in Berkshire (England) → Wessex Archaeology.
Bell Beaker rich lady's burial unearthed in Berkshire (England) → Wessex Archaeology.
The middle-aged woman wore a necklace of tubular golden beads, amber buttons on her clothes and a possible lignite bracelet. She was accompanied by a bell-shaped beaker of the "corded" type (oldest and roughest variant, of likely Central European inception).
The chemical signature of the gold beads is coherent with deposits from Southern Britain and SE Ireland.
Giza pyramid construction's logistics revealed → Live Science.
Caesar beat the Gauls.Was there not even a cook in his army?
Bertolt Brecht (A Worker reads History)
Now we know that at the very least the famed early pharaohs Khafra, Khufu and Menkaure, who ordered the massive pyramids of Giza to be built as their tombs did have some cooks in charge of feeding the many workers who actually built them, stone by stone.
These workers were housed in a village some 400 meters south of the Sphinx, known as Heit el-Ghurab. In this place archaeologists have found a cemetery, a corral with apparent slaughter areas and piles of animal bones. Based on these, researchers estimate that more than 2,000 kilograms of meat were eaten every day during the construction of Menkaure's pyramid, the last and smallest one of the three geometric mounds.
The figures estimated for such a logistic operation border disbelief: 22,000 cows, 55,000 sheep and goats, 1200 km² of grazing land (roughly the size of Los Angeles or 5% of the Nile Delta), some 3500 herders (adding up to almost 20,000 people if we include their families).
A curious detail is that most of the beef was destined to the building of the overseers, while the common workers were mostly fed sheep or goat instead. Another settlement to the East of apparently local farmers ate most of the pork. There were also temporary tent camps closer to the pyramids.
Iron Age
Late Indus Valley Civilization was overcome by violence → National Geographic.
 |
| Harappa (CC by Shephali11011) |
The Late Indus Valley Civilization (Cemetery H cultural layer, usually attributed to the Indoeuropean invasions) was, unlike in previous periods, quite violent, new evidence highlights.
The evidence from the bones also highlights the arrival of many non-local men, who apparently married local women. But the most shocking element is the striking evidence of widespread violence:
The skull of a child between four and six years old was cracked and crushed by blows from a club-like weapon. An adult woman was beaten so badly—with extreme force, according to researchers—that her skull caved in. A middle-aged man had a broken nose as well as damage to his forehead inflicted by a sharp-edged, heavy implement. Of the 18 skulls examined from this time period, nearly half showed serious injuries from violence ...
Gaming pieces of Melton Mowbray (England) → Science Daily.
Excavation of a hillfort at Burrough Hill revealed ancient gaming pieces, among other materials.

 |
| (CC by P.A. Salguero Quiles) |
The tomb has an access gate and is estimated to be from the 5th or 4th centuries BCE (Iberian culture) and, unlike most burials of the time, the corpse was not incinerated.
The finding highlights the need for further archaeological work in all the hill but the severe budgetary cuts threaten this development.
Baza (Granada) hosts a dedicated archaeological museum inaugurated in 2011.
Tocharian mummy buried with marijuana hoard → Paleorama[es].

Some 800 grams of the psychedelic plant, including seeds, were found at the burial place of a Tocharian man, presumably a shaman, at Yanghai (Uyghuristan), belonging to the Gushi culture and dated to at least 2700 years ago. The plant belongs to a cultivated variety.
Some of the oldest cannabis evidence are also from that area (Pazyrk culture c. 2500 years ago) and also from Nepal (Mustang, similar dates). Later in Southern Central Asia it was used in combination with opium and ephedra, from where soon migrated to South Asia and many other parts of Eurasia.
Genetics

New device radically reduces costs and time in DNA extraction → Science Daily.
Researchers from the University of Washington and NanoFacture Inc. have developed a device, which looks like a kitchen appliance, able to extract DNA from tissues (like saliva or blood) in minutes at low cost and without using the toxic chemicals habitual in the field.
The prototype is designed for four samples but can be scaled for the lab standard of 96 samples at once.
March 16, 2013
Plants do use epigenetics to adapt to diverse environments
If you've ever grown plants that come from a distant land, you may be familiar with the fact that you may need an adaptive process of one or more generations to get the best of them. Oddly enough this time is often too short for genetic adaption to happen and sweep over, especially as most of the ill-adapted plants don't really die nor fail to reproduce (i.e. they are not too aggressively selected against). How does it happen then? Epigenetics may have the answer.
Robert J. Schmidt et al., Patterns of population epigenomic diversity. Nature 2013. Pay per view → LINK [doi:10.1038/nature11968]
Abstract
Natural epigenetic variation provides a source for the generation of phenotypic diversity, but to understand its contribution to such diversity, its interaction with genetic variation requires further investigation. Here we report population-wide DNA sequencing of genomes, transcriptomes and methylomes of wild Arabidopsis thaliana accessions. Single cytosine methylation polymorphisms are not linked to genotype. However, the rate of linkage disequilibrium decay amongst differentially methylated regions targeted by RNA-directed DNA methylation is similar to the rate for single nucleotide polymorphisms. Association analyses of these RNA-directed DNA methylation regions with genetic variants identified thousands of methylation quantitative trait loci, which revealed the population estimate of genetically dependent methylation variation. Analysis of invariably methylated transposons and genes across this population indicates that loci targeted by RNA-directed DNA methylation are epigenetically activated in pollen and seeds, which facilitates proper development of these structures.
From the body of the article:
Epiallele formation in the absence of genetic variation can result in phenotypic variation, which is most evident in the plant kingdom, as exemplified by the peloric and colorless non-ripening variants from Linaria vulgaris and Solanum lycopersicum, respectively6, 7. Although rates of spontaneous variation in DNA methylation and mutation can be decoupled in the laboratory8, 9, 10, 11, in natural settings, these two features of genomes co-evolve to create phenotypic diversity on which natural selection can act.
...
Similarly to the limited examples of pure epialleles (methylation variants that form independent of genetic variation), few examples of DNA methylation variants linked to genetic variants are known15, 16, 17.
And the 'Conclusion remarks' (emphasis is mine):
Natural epigenomic variation is widespread within A. thaliana, and the population-based epigenomics presented here has uncovered features of the DNA methylome that are not linked to underlying genetic variation, such as all forms of SMPs and CG-DMRs. However, C-DMRs have positional association decay patterns similar to linkage disequilibrium decay patterns for SNPs and in some cases are associated with genetic variants, but the majority of C-DMRs were not tested by association mapping due to low allele frequencies and could result from rare sequence variants. Our combined analyses of genetic and methylation variation did not uncover a correlation between major effect mutations and genes silenced by RdDM, suggesting that this pathway may target these genes for another purpose. This purpose could be to restrict expression from vegetative tissues similarly to transposons. Another possible purpose of being targeted by RdDM could be to coordinate expression specifically in pollen and in seed to ensure proper gametophytic and embryonic development. Animals also use small RNA-directed DNA methylation and heterochromatin formation mechanisms to maintain the epigenome of the germ line through the use of Piwi-interacting RNAs36. In both plants and animals these small RNAs are derived from the genome of companion cells, which are terminal in nature and can afford widespread reactivation of transposon and repeat sequences as they are not passed on to the next generation. Our study provides evidence that RdDM-targeted genes may have co-opted this transposon silencing mechanism to maintain their silenced state in vegetative tissues and transgenerationally, as well as to ensure proper expression important for pollen, seed and germ line development.
March 2, 2013
Non-additive genetic models and the problem of stasis (relative stability of the genomes) and missing heritability
A simple and classical approach to genetic understanding of phenotype variability is to assume that allele influence is additive. However this may be just a mirage of imperfect methodology.
In any case the additive model approach has reached a point when it is quite obvious not enough to explain the genetic background of phenotype heritability.
Gibran Hemani et al., An Evolutionary Perspective on Epistasis and the Missing Heritability. PLoS Genetics 2013. Open access → LINK [doi:10.1371/journal.pgen.1003295]
AbstractThe relative importance between additive and non-additive genetic variance has been widely argued in quantitative genetics. By approaching this question from an evolutionary perspective we show that, while additive variance can be maintained under selection at a low level for some patterns of epistasis, the majority of the genetic variance that will persist is actually non-additive. We propose that one reason that the problem of the “missing heritability” arises is because the additive genetic variation that is estimated to be contributing to the variance of a trait will most likely be an artefact of the non-additive variance that can be maintained over evolutionary time. In addition, it can be shown that even a small reduction in linkage disequilibrium between causal variants and observed SNPs rapidly erodes estimates of epistatic variance, leading to an inflation in the perceived importance of additive effects. We demonstrate that the perception of independent additive effects comprising the majority of the genetic architecture of complex traits is biased upwards and that the search for causal variants in complex traits under selection is potentially underpowered by parameterising for additive effects alone. Given dense SNP panels the detection of causal variants through genome-wide association studies may be improved by searching for epistatic effects explicitly.
It is difficult for me to explain (or even understand well in some aspects) the issues under debate here but some excerpts from the text do ring very true in my mind:
There exists a paradox in evolutionary biology. Despite a near-ubiquitous abundance of genetic variation [1] traits under selection often evolve more slowly than expected and, contrary to expectation, genetic variation is maintained under selection. This problem is known as ‘stasis’ [2], [3], and it is particularly evident in fitness-related traits where the genetic variation tends to be highest [4] yet there is commonly no observed response to selection at all [5]–[7]....After hundreds of genome-wide association (GWA) studies [11] a picture is emerging where the total genetic variation explained by variants that have been individually mapped to complex traits is vastly lower than the amount of genetic variation expected to exist as estimated from pedigree-based studies, a phenomenon that has come to be known as the problem of the ‘missing heritability’ [12]. Again, there are probably numerous contributing factors, and ostensibly the most parsimonious explanation is that complex traits comprise many small effects that GWA studies are underpowered to detect [13], [14], but whether this is the complete story deserves exploration....Beyond the realm of complex trait genetics it appears that epistasis does appear to be common. For example in molecular studies it is routine to observe ‘phenotypic rescue’ where the phenotypic effect of a gene knockout can be masked by a second knockout (e.g. [31]). Another commonly encountered form of epistasis is ‘canalisation’ [32], where phenotypes exhibit robustness to the knockout of one gene, requiring a second knockout to elicit a response (e.g. [33]). At the macroevolutionary scale, epistasis is also of central importance, for example it has recently been shown that an advantageous substitution in one species is often found to be deleterious in others, thus the substition's effect on fitness is dependent upon the genetic background in which it is found [34]....The results suggest that we should expect significant levels of non-additive variation to be maintained in fitness-related traits.
One of the criticisms seems to suggest that studying homogeneous populations in GWAS is inefficient because linkage disequilibrium (LD) masks the non-additive effect of the alleles, making them appear, erroneously, as additive.
... it was observed that even with modest reductions in LD between causal variants and observed SNPs all testing strategies tended to decline in performance rapidly....The example of the single locus case, overdominance, is central to processes of heterosis and inbreeding depression [52], [53], and has been identified in molecular studies also [54], [55]. Indeed, heterozygote advantage plays an important role in evolutionary theory, as it confers segregational load on a population, and this type of load cannot be purged due to balancing selection, potentially rendering populations susceptible to accumulating a critical mass of such polymorphisms [56]....It is important to note that the processes underlying stasis and missing heritability are unlikely to be caused by any single factor. For example, a compelling argument is that though most traits exhibit genetic variation, selection acts upon multidimensional trait space in which there is no genetic variation [59], and this will hold under an additive model of genetic variation.
December 21, 2012
Hominid speciation: sudden or gradual?
It depends apparently: bonobos may have diverged quite suddenly while in other cases, including the Pan-Homo split, the process of speciation appears to have been more gradual.
Thomas Mailund et al., A New Isolation with Migration Model along Complete Genomes Infers Very Different Divergence Processes among Closely Related Great Ape Species. PLoS ONE 2012. Open access → LINK [doi:10.1371/journal.pgen.1003125]
Abstract
We present a hidden Markov model (HMM) for inferring gradual isolation between two populations during speciation, modelled as a time interval with restricted gene flow. The HMM describes the history of adjacent nucleotides in two genomic sequences, such that the nucleotides can be separated by recombination, can migrate between populations, or can coalesce at variable time points, all dependent on the parameters of the model, which are the effective population sizes, splitting times, recombination rate, and migration rate. We show by extensive simulations that the HMM can accurately infer all parameters except the recombination rate, which is biased downwards. Inference is robust to variation in the mutation rate and the recombination rate over the sequence and also robust to unknown phase of genomes unless they are very closely related. We provide a test for whether divergence is gradual or instantaneous, and we apply the model to three key divergence processes in great apes: (a) the bonobo and common chimpanzee, (b) the eastern and western gorilla, and (c) the Sumatran and Bornean orang-utan. We find that the bonobo and chimpanzee appear to have undergone a clear split, whereas the divergence processes of the gorilla and orang-utan species occurred over several hundred thousands years with gene flow stopping quite recently. We also apply the model to the Homo/Pan speciation event and find that the most likely scenario involves an extended period of gene flow during speciation.
October 23, 2012
Blood groups A and B inherited from simian ancestors
A new study has found that blood group A and B alleles have been stable (albeit in likely dynamic equilibrium) in the primate family since... always.
Abstract
The ABO histo-blood group, the critical determinant of transfusion incompatibility, was the first genetic polymorphism discovered in humans. Remarkably, ABO antigens are also polymorphic in many other primates, with the same two amino acid changes responsible for A and B specificity in all species sequenced to date. Whether this recurrence of A and B antigens is the result of an ancient polymorphism maintained across species or due to numerous, more recent instances of convergent evolution has been debated for decades, with a current consensus in support of convergent evolution. We show instead that genetic variation data in humans and gibbons as well as in Old World monkeys are inconsistent with a model of convergent evolution and support the hypothesis of an ancient, multiallelic polymorphism of which some alleles are shared by descent among species. These results demonstrate that the A and B blood groups result from a trans-species polymorphism among distantly related species and has remained under balancing selection for tens of millions of years—to date, the only such example in hominoids and Old World monkeys outside of the major histocompatibility complex.
Razib has some more details on the matter (being PPV, I haven't read it). Still he wonders what kind of disease or otherwise evolutionary pressure may have been so virulent as to keep the whole order of primates (or at the very least all simians) on our toes all these millions of years.
The answer may well be known already: it seems that type A blood protects against the plague, while type B protects against smallpox, both great historical killers of those without enough defenses. However they may favor other less important health problems like blood clots or cancer, enough to exert a mild pressure in favor of a return to the basic type zero ("O"), which would have evolved by loss of function once and again.
Whatever the case I find fascinating that these immune mechanisms may be so extremely persistent and I wonder if the bacterian mechanisms they confront may be more generic than just an specific disease.
See also: maps of distribution of major blood types.
Update: a pre-print copy of the paper is available at arXiv.
Update: a pre-print copy of the paper is available at arXiv.
September 12, 2012
Convergent evolution towards big brains in humans and dolphins

Every whale and dolphin evolved from a deer-like animal with slender, hoofed legs, which lived between 53 and 56 million years ago. Over time, these ancestral creatures became more streamlined, and their tails widened into flukes. They lost their hind limbs, and their front ones became paddles. And they became smarter. Today, whales and dolphins – collectively known as cetaceans – are among the most intelligent of mammals, with smarts that rival our own primate relatives.
Now, Shixia Xu from Nanjing Normal University has found that a gene called ASPM seems to have played an important role in the evolution of cetacean brains. The gene shows clear signatures of adaptive change at two points in history, when the brains of some cetaceans ballooned in size. But ASPM has also been linked to the evolution of bigger brains in another branch of the mammal family tree – ours. It went through similar bursts of accelerated evolution in the great apes, and especially in our own ancestors after they split away from chimpanzees.
It seems that both primates and cetaceans—the intellectual heavyweights of the animal world—could owe our bulging brains to changes in the same gene. “It’s a significant result,” says Michael McGowen, who studies the genetic evolution of whales at Wayne State University. “The work on ASPM shows clear evidence of adaptive evolution, and adds to the growing evidence of convergence between primates and cetaceans from a molecular perspective.”
... continue reading at Not Exactly Rocket Science.
Note: it is one of the microcephalin genes, if you wondered.
September 6, 2012
ENCODE: from mere protein-coding to true program-like understanding of the human genome

It is in the news these days: an intercontinental army of some 440 researchers have taken decisive steps to truly understand the human genome, our base program, helped by the much lower costs of genome sequencing achieved recently.
One of the most remarkable results is discarding that most of our genome is "junk DNA". Until recently many though that only some 20% of the genome, the protein-coding segments, were meaningful, while the rest was useless "junk" mysteriously accumulated through the millennia.
Nature is much more efficient than that, it seems, and the reality is that the remaining 80% of the genome is a maze of switches that actually regulate how cells, and the whole body, are built and maintained. A true biological program encoded in DNA.
The main product of this intercontinental effort is a threaded encyclopedia of the human genome, as well as three (freely accessible) articles in Nature:
- Presenting ENCODE (M. Skipper, R. Dhand, P. Campbell)
- Genomics: ENCODE explained (Joseph R. Ecker et al.)
- An integrated encyclopedia of DNA elements in the human genome (ENCODE Project Consortium)
In addition to all this you may want to visit the ENCODE site or read this one or this other article at SD, for example.
August 24, 2012

August 23, 2012
Increased complexity in certain regions sets apart human and chimp brains
 |
| Frontal lobe (CC-BY-SA-2.1-jp) |
{kind=link}
This paper looks like a very important research piece for the understanding of the human mind, of what makes our brains specifically human and ultimately of what makes ourselves what we are.
Genevieve Konopka et al., Human-Specific Transcriptional Networks in the Brain. Neuron 2012. (Freely accessible apparently) ··> LINK [doi:10.1016/j.neuron.2012.05.034]
Summary
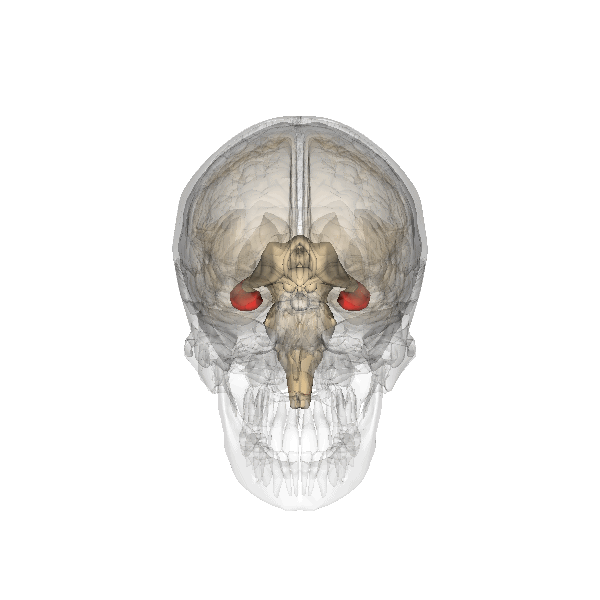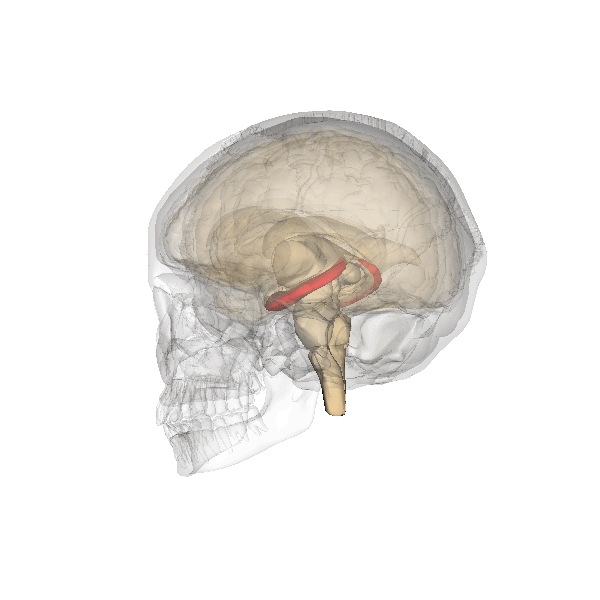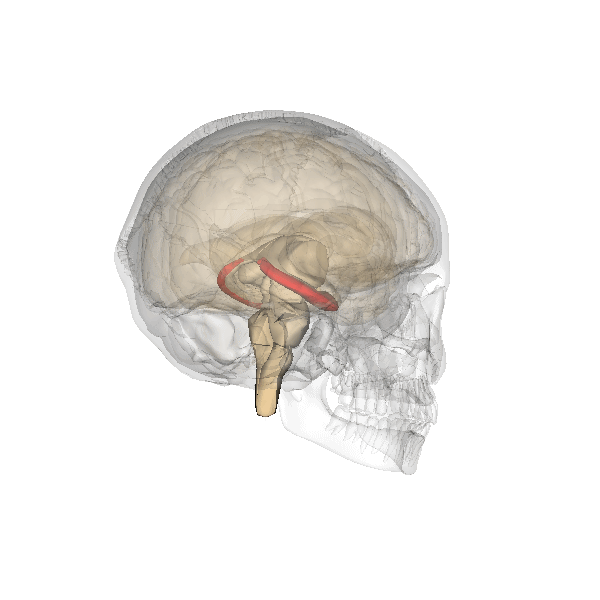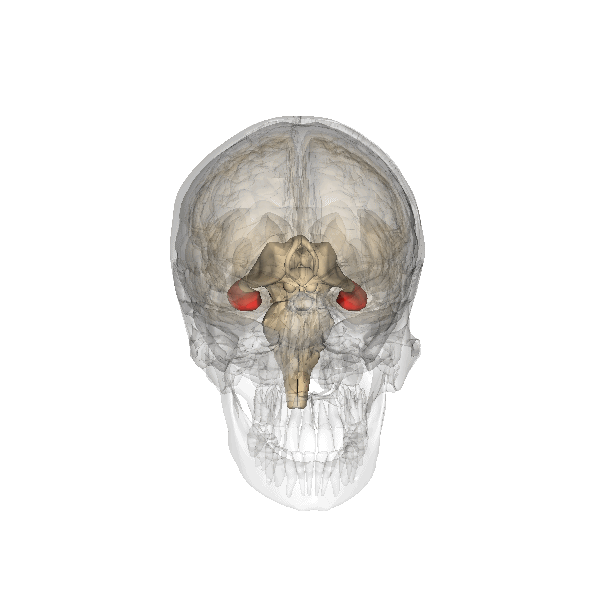
Understanding human-specific patterns of brain gene expression and regulation can provide key insights into human brain evolution and speciation. Here, we use next-generation sequencing, and Illumina and Affymetrix microarray platforms, to compare the transcriptome of human, chimpanzee, and macaque telencephalon. Our analysis reveals a predominance of genes differentially expressed within human frontal lobe and a striking increase in transcriptional complexity specific to the human lineage in the frontal lobe. In contrast, caudate nucleus gene expression is highly conserved. We also identify gene coexpression signatures related to either neuronal processes or neuropsychiatric diseases, including a human-specific module with CLOCK as its hub gene and another module enriched for neuronal morphological processes and genes coexpressed with FOXP2, a gene important for language evolution. These data demonstrate that transcriptional networks have undergone evolutionary remodeling even within a given brain region, providing a window through which to view the foundation of uniquely human cognitive capacities.
 |
| Hippocampus (CC-BY-SA-2.1-jp) |
{kind=link}
For what I could understand, mostly from the press release, the authors unveiled increased complexity of the gene expression modulating three regions of our brains: the frontal cortex, the hippocampus and the striatum.
It is not a mere matter of size but specially one of much increased complexity in the wiring of these three regions what seems to make our brains unique.
The research also reinforces the apparent importance of the much debated genes CLOCK (affecting circadian rhythms, mood, pregnancy and metabolism), FOXP1 and FOXP2 (related specially with speech), whose connectivity is much increased in humans in comparison with our ape cousins.
June 18, 2012
Denisovan and Neanderthal proviral DNA
A provirus is a strand of autosomal DNA that was inserted by a virus once upon a time and got lost in our genome as junk DNA, not being anymore active (would it remain active it'd be a retrovirus). Such insertions are thought to be unique phylogenetic events.
New research has identified a provirus* (HERV-K-Ne1 = HERV-K-De6, inserted in Chromosome 5) shared by Neanderthals and Denisovans but not Homo sapiens. This is consistent with the previous data that placed their autosomal DNA closer to each other than to Homo sapiens.
Lorenzo Agoni et al., Neandertal and Denisovan retroviruses. Current Biology, 2012. Freely accessible (letter with supplementary material) at the time of writing this.
It must be noted however the mitochondrial DNA, inherited by pure matrilineage, is much closer among our species and Neanderthals than either one with Denisovans, what to me suggest that Denisovans are no new species but a hybrid of Neanderthal and Homo erectus. A theory not yet fully testable for lack of DNA from Asian Homo erectus.
Interestingly Denisovans have also several proviruses not found in Neanderthals, what could well support my theory of hybridization. The detected provirus could hence have migrated from Neanderthals to Denisovans in the hybridization episode (along with lots of other autosomal DNA), while the rest could have been retained from the H. erectus ancestors by the maternal line.
However as the article is both very technical and succinct, I can't be sure right now of how strongly or weakly can this info support the hybridization model (founded opinions welcome).
In total the researchers detected three Neanderthal proviruses and 12 Denisovan ones, one of which is shared between both nominal species. It is convenient to remind that while the Denisovan genome was very well preserved and sequenced almost completely, the Neanderthal genome is only known in fragmentary form, amounting to about 60% of the actual genome.
June 14, 2012
Bonobo genome sequenced
 |
| Ulundi (source) |
The last great ape* to be sequenced has been the bonobo, it complements the Homo sapiens, Neanderthal, Denisovan (probably a hybrid), chimpanzee, gorilla and orangutan genomes:
Kay Prüfer et al., The bonobo genome compared with the chimpanzee and human genomes. Nature 2012. Open access.
The reference genome was sequenced from a female bonobo captive at Leizpig Zoo, known as Ulundi.
The genome will, hopefully, help understand better the genetic basis of our being as humans and, maybe also get some inferences on our prehistory.
Stubbornly under-estimating divergence times by almost 100%
In this sense I want to emphasize that the paper insists in producing Pan-Homo and internal Pan divergence times that are irrationally low. The cause of this systematic error that persists through some literature seems to be rooted on the Homo-Pongo divergence estimate, which I do not know the details about but seems from context to be an extreme under-estimate.
The matter was already debated in 2008 by Jenniffer L. Caswell, who explained that the Bonobo-Chimpanzee split cannot be more recent than 1.5 to 2.0 million years because it was then when the Congo River was formed separating the two populations radically (allopatric speciation). This is quite apparent in the distribution of bonobos and chimpanzees:
 |
| fig. 1a |
So unless the geology is wrong, bonobos and chimpanzees diverged 1.5 to 2 million years ago, and not a mere million years ago, as this paper claims.
This has important implications for the Homo-Pan divergence age, as I have discussed again and again. Assuming that the 4.5:1 ration estimated in this paper is correct, then the actual Homo-Pan divergence age ranges between 6.8 to 9.0 million years ago (and not a mere 4.5 Ma), with a median of 7.9 Ma, quite similar to the 8 Ma estimate I have been defending since the Caswell paper was published in 2008.
See also
- Category: bonobo at my old blog Leherensuge
- Category: bonobo at this blog
- Category: human evolution
- Molecular clock obscuring the real origin of primates
- Chimps and humans diverged 8 million years ago
_________________________________________________________________________
* Note: I know someone will say that Homo sp. are not "apes" but I say Homo are a subset of the great apes clade (Hominidae) phylogenetically and therefore great apes ourselves - something to be irrationally proud of, of course.
June 12, 2012
Concern for the use of genetic tests for Nazi purposes
As Van Ardsdale explains purity is not a genetic reality, first of all because each time a new person is conceived (by the enjoyable but quite impure act of sex) admixture takes place (and if mum and dad are genetically too similar, then inbreeding happens what is generally bad). So whoever would wish to imagine themselves as pure should not look into genetics but into Platonic solids or something.
But the right tools in the wrong hands typically has the wrong results. And the tool of genetic analysis in the hands of Hitler* or the like could be used to entice racist discrimination.
Nature reports that a Hungarian genetic testing company, Nagy Gén, has issued a certificate by which a person, a Hungarian Nazi member of the criminal Jobbik party, was said to have no Jewish nor Roma ancestry.
Nagy Gén scanned 18 positions in the MP’s genome for variants that it says are characteristic of Roma and Jewish ethnic groups; its report concludes that Roma and Jewish ancestry can be ruled out.
It's difficult to imagine how the company could certify that because there are no absolute lines defining such ethnic categories, not in the genetic aspect either, just clinal trends.
I understand from the context (18 positions) that the test is one of those biometric AIM-based tests that police uses sometimes to attempt to guess (without any certainty) the ancestry of suspects.
The scandalous certificate was first posted at a Nazi site, which praised the intent but (correctly) dismissed the scientific quality of the test. It was later republished at a Magyar-language news blog.
The affair underlines the dangers of all kind of biometrics, be them genetic or anthropometric, when used for reasons that are not pure science. That's a reason why I do not generally favor private, commercial genetic testing but rather academic studies of populations with prehistory reconstruction intent.
Personally I have never got myself tested nor I really care much because what matter for me is not "my" private ancestry but, if anything, the ancestry of the diverse peoples and communities, what can tell us something about their history and prehistory.
______________________________
* Incidentally, I suspect that Hitler would have got serious problems promoting his racist ideas if he would have got access to genetic analysis because his paternal lineage was quite Mediterranean and ultimately rooted in Africa (it could even be Jewish, although hard to tell ultimately). He would have had to lie even to himself, abandon his racism or maybe kill himself (mostly good results).
March 8, 2012
Gorilla genome sequenced
 |
| Kamilah the gorilla |
The full genome of Kamilah, a female gorilla from San Diego zoo, has been sequenced. With this one all extant great apes, excepted the bonobo, have been fully sequenced.
A. Scally et al., Insights into hominid evolution from the gorilla genome sequence. Nature 2012 (open access).
The authors propose a divergence of Gorilla from the Pan-Homo branch c. 10 million years ago. But this is based on a most unlikely assumption of Pan-Homo divergence happening only 6 million years ago, when it's surely of at least 8 million years (and maybe as many as 10 million). A corrected estimate for the Gorilla branch could then be between 13 to 18 million years in fact.
See on this regard:
Subscribe to:
Posts (Atom)